


【中睿行业分享】中国创新药出海

免责声明:本文内容仅供读者参考,不作为任何投资建议;股市有风险,投资需谨慎。
01
为什么要出海
随着国内新药研发实力的快速提升,药物创新性和研发效率的优势逐步受到全球的认可,新药出海本来可以循序渐进慢慢做,但在国内带量采购、医保谈判、创新药内卷加剧等冲击下,寻找新的市场和新的发展空间,对于国内创新药企业来说,变得越发急迫。
此外,由于社会体制和市场发展阶段不同,相对成熟和资本化的欧美市场在创新药产品定价、研发资金支持、配套制度等方面对药企出海具有明显吸引力。
(一)支付能力差
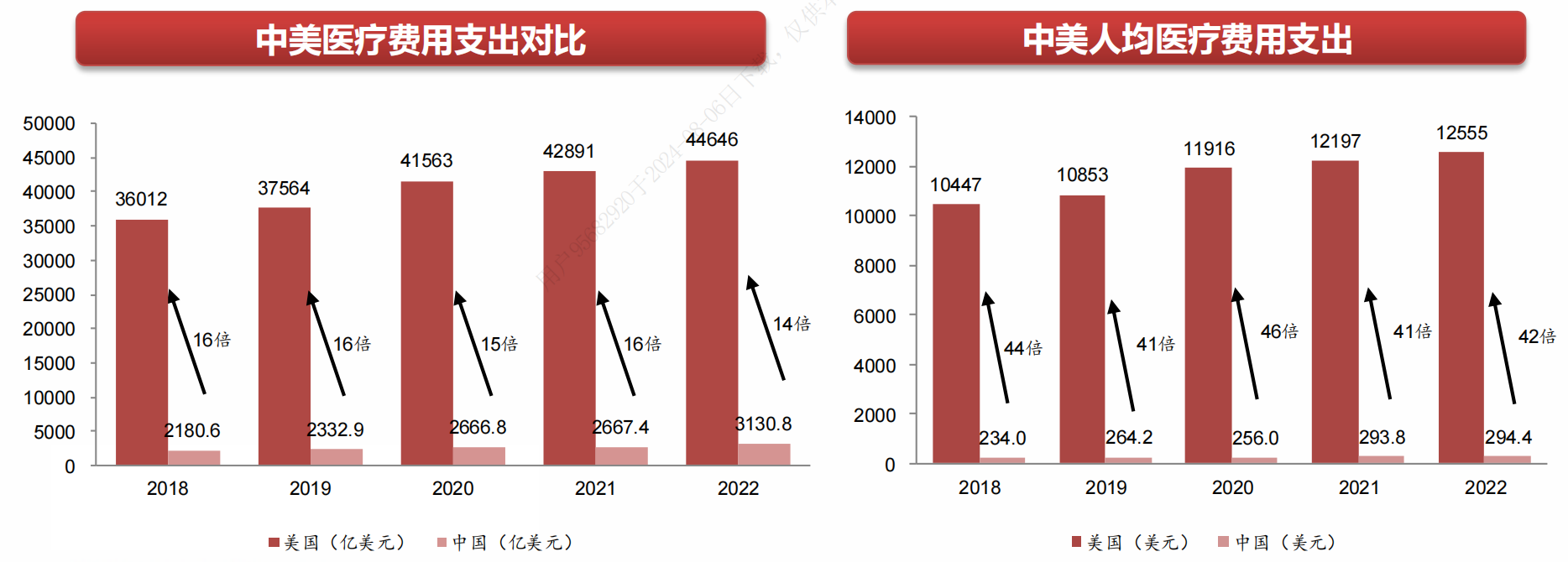
我国人口众多,医疗市场体量庞大,位列全球第二,仅次于美国,但是第一和第二的差距比较夸张。
2018年,美国总医疗费用支出是中国的16倍,同年美国人均医疗费用支出是中国的44倍。尽管近年来中国医疗行业发展迅速,差距逐步缩小, 2022年美国医疗费用支出是中国的14倍,人均医疗费用是中国的42倍。
(二)药品定价差距
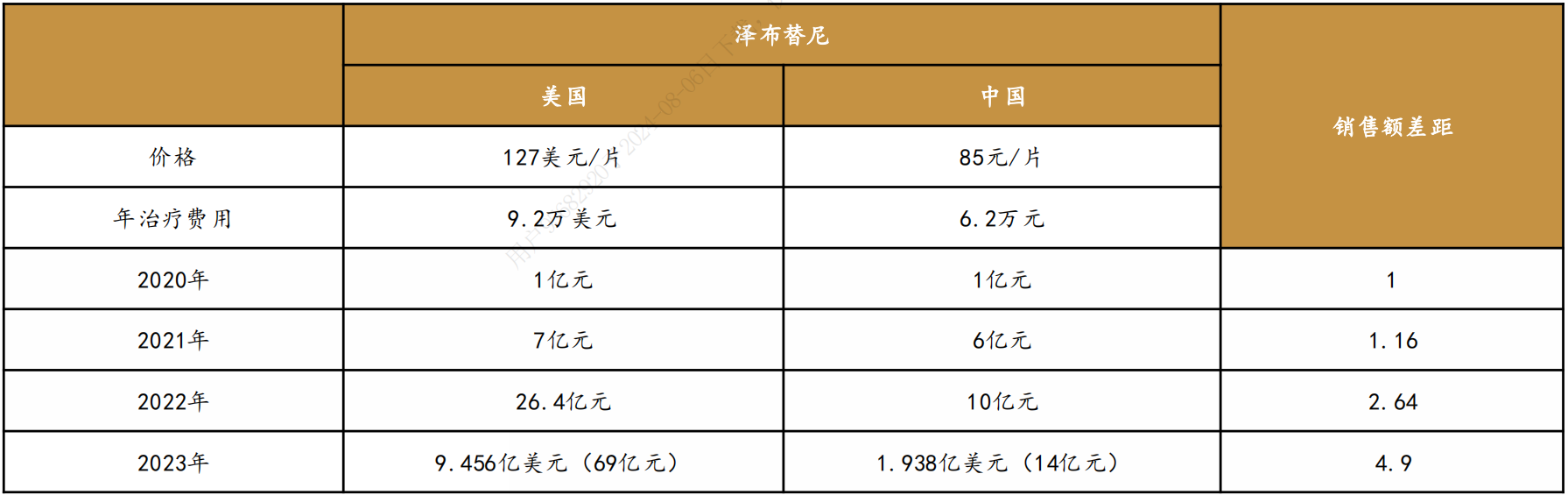
以百济神州的泽布替尼为例,2022年,泽布替尼在美国实现26.4亿元销售额,国内销售额为10亿元。2023年,泽布替尼美国收入69亿元,中国收入14亿元,相差4.9倍。这背后深刻体现了中美两国在定价、渠道推广、销售峰值等多项差异。
君实生物的PD-1单抗于2023年10月底获得美国FDA的批准后,在美国的定价为每瓶8892.03美元,折合人民币约63604.69元,这一价格是其在中国市场不足2000元人民币售价的30倍以上。
同样,和黄医药的呋喹替尼于2023年11月初获得美国FDA批准后,在美国市场的定价为25200美元,折合人民币约18.04万元,这是其在中国市场7500多元人民币售价的24倍。

(三)资金回笼差异
随着近几年生物医药行业投融资下降,IPO步伐减缓,在高研发投入不变甚至提升的大背景下,部分创新药企业账上资金越发紧张,“活下去”成为创新药企业追求的目标之一。
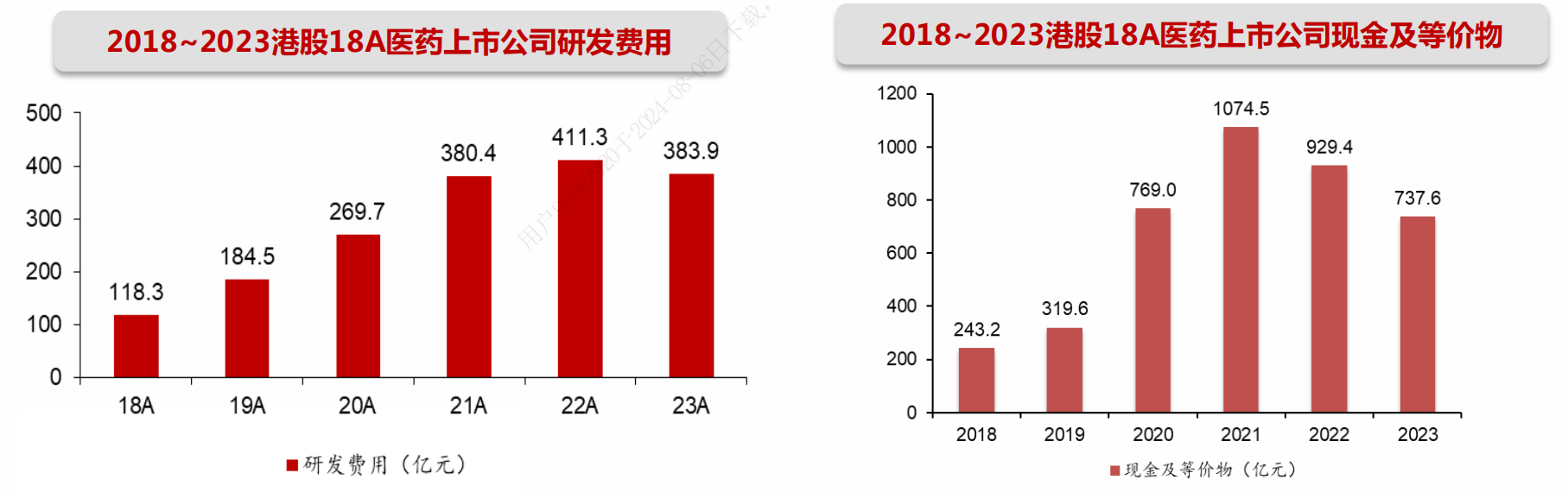
而创新药出海,尤其是License-out,首付款可迅速回笼资金。此外,海外临床研究,尤其是Ⅲ期临床研究,研发成本高昂,通过License-out授权,可将海外临床研发成本交由海外企业承担。
对于尚未有产品商业化收入的科创板Biotech,“现金/研发费用“比值中位数约为4.9,而好产品能够获得的授权收入等可以迅速贡献大量现金,比如百利天恒旗下产品于2023年底和BMS的重磅合作,获得8亿美元的首付款和最高5亿美元的近期付款。
非科创板的A股药企基本具备较稳定的自我造血能力,“经营现金流量额/研发费用”比值中位数约为1.8。
港股创新药公司“现金等/研发费用“比值中位数约为2.9,其中100亿以上的pharma型公司均具有较充裕的现金和自身造血能力;Biotech随着管线陆续步入收获期以及出海,已经有近半数公司产生收入。
因此,对于中国创新药企业而言,不但大幅减少了资金压力,更有助于快速推进临床试验。
02
靠什么出海
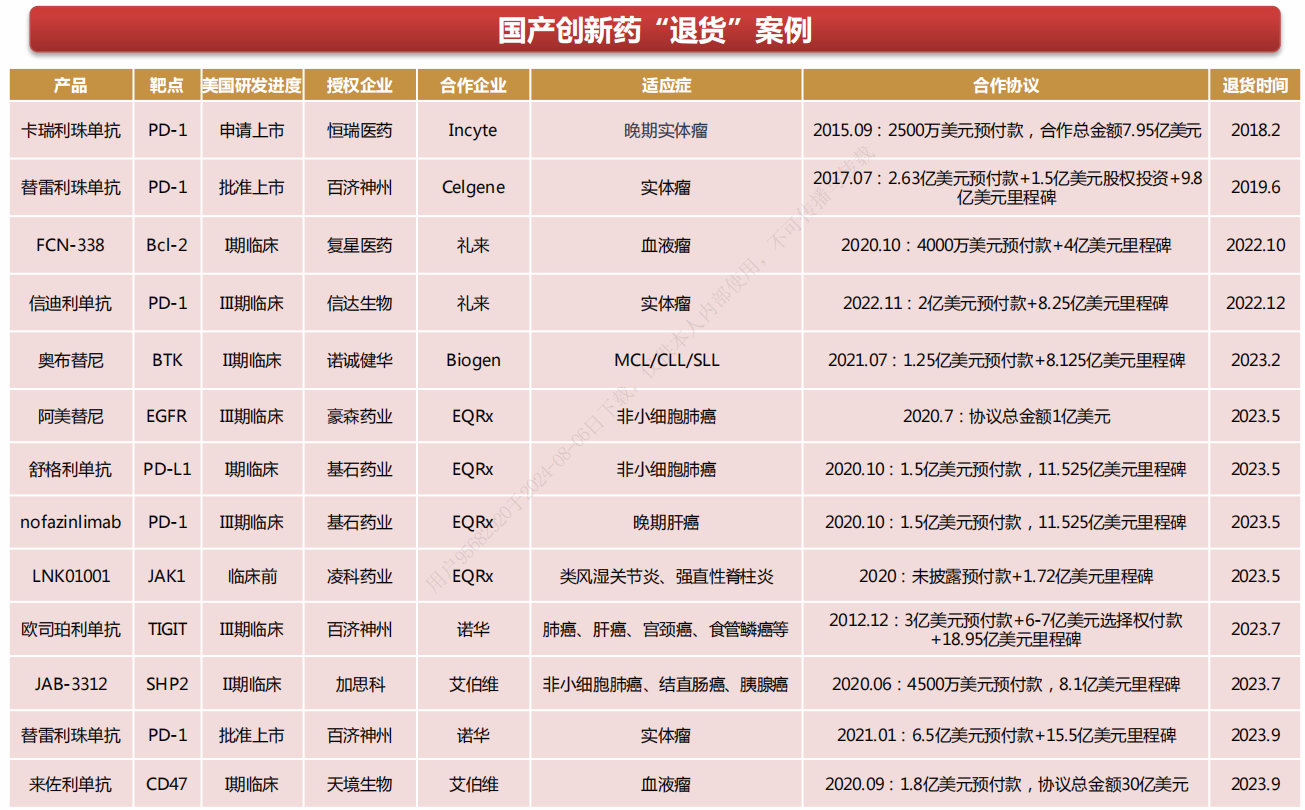
近年来,已经有超50款国产创新药成功出海,并且多款都实现了10亿以上的成交金额。但必须承认的是,国产创新药海外授权从来不是一件容易的事情,尤其是10亿以上的大单,受阻退货的也不少。
从受阻原因来看,基本都是缺乏代表美国患者人群的国际多中心临床试验数据或者海外临床试验疗效不及预期。

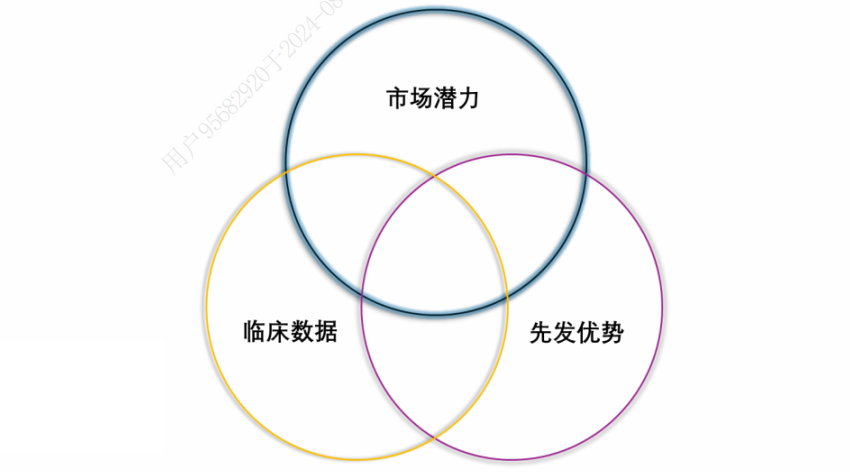
因此,要想顺利出海并实现项目的顺利推进,至少需满足下面三要素:
首先,市场空间巨大,可有效解决未满足的临床需求。市场空间的潜力是MNC(跨国药企)最在意的问题,比如强生携手传奇生物,强生预计该产品全球销售额有望达50亿美元。Summit Therapeutics引进康方生物的AK112,也是考虑到K药2023年全球销售额达250亿美元。
其次,临床有效性及安全性较目前标准疗法优势明显。药物的临床疗效和安全性优势是药品拓展市场的关键,最终反映出医生和患者对药品的信赖度。武田医药引进和黄的呋喹替尼,一方面是呋喹替尼疗效优势明显,另一方面是呋喹替尼安全性良好,安全性的优势意味着后续还能探索更多适应症或者联合用药方案。
最后,临床进展居前,先发优势明显。药品的先发优势对于药物的放量至关重要,这一点在海内外都是一样的。根据《2020年度中国抗肿瘤新药临床研究评述》,全球范围内同一靶点药物,首个上市产品可以获得45%的市场份额,第二至第四个上市的产品分别可以获得27.9%、14%以及11.3%的市场份额。
03
什么方式出海
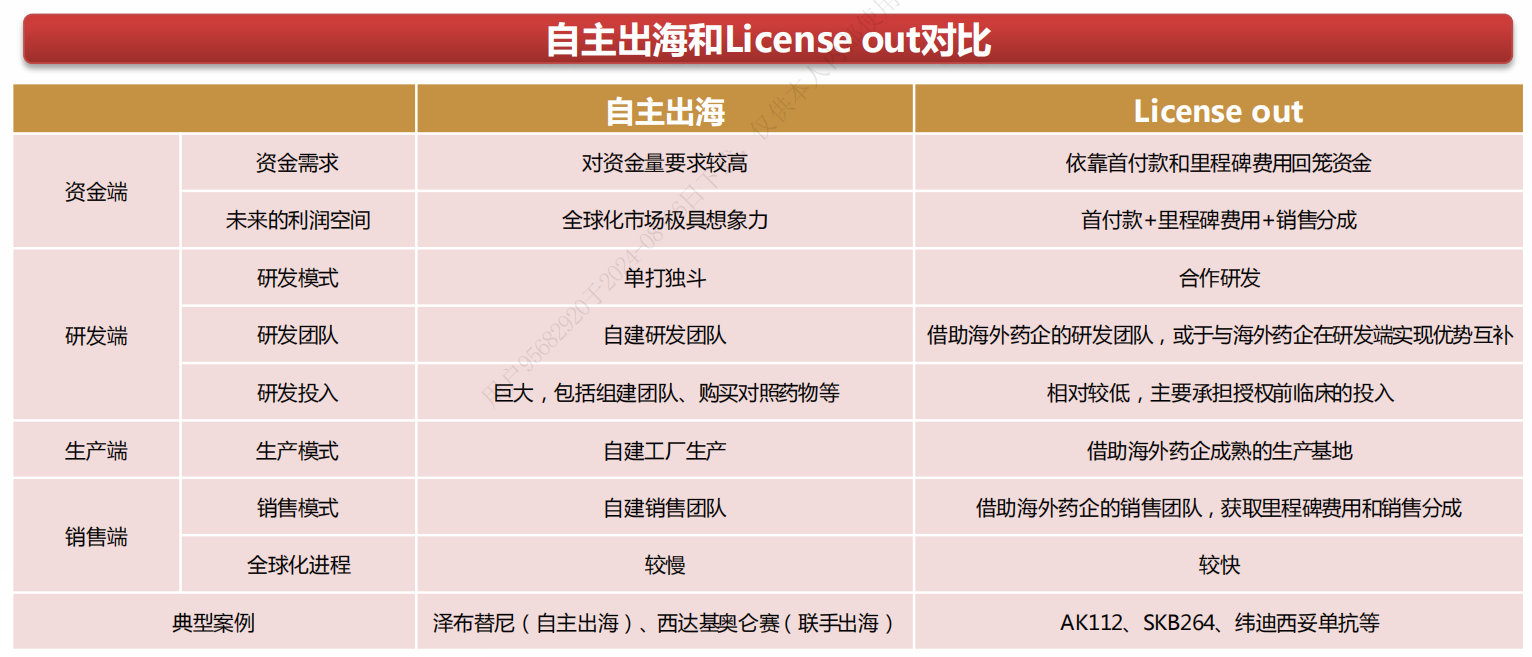
出海主要分为自主出海、License-out(借船出海)和联手出海。
自主出海即中国药企凭借自身的团队在海外国家和地区开展临床试验,申报上市,获批后展开销售。考虑到自主出海的高风险,部分企业采用联手出海的模式,即通过与海外药企联合开发,分担成本并分享收益。
License-out则是中国药企将自身产品的海外权益或全球权益出售许可给以欧美跨国药企为代表的制药企业,获得首付款和里程碑费用。海外药企接过接力棒后,负责海外市场的临床开发、申报上市、生产及销售工作。
目前License-out是出海的主要模式,企业可根据自身的战略规划和实力,在不同阶段选择不同的模式,或采用多种模式并行的方式。
(一)自主出海&联手出海
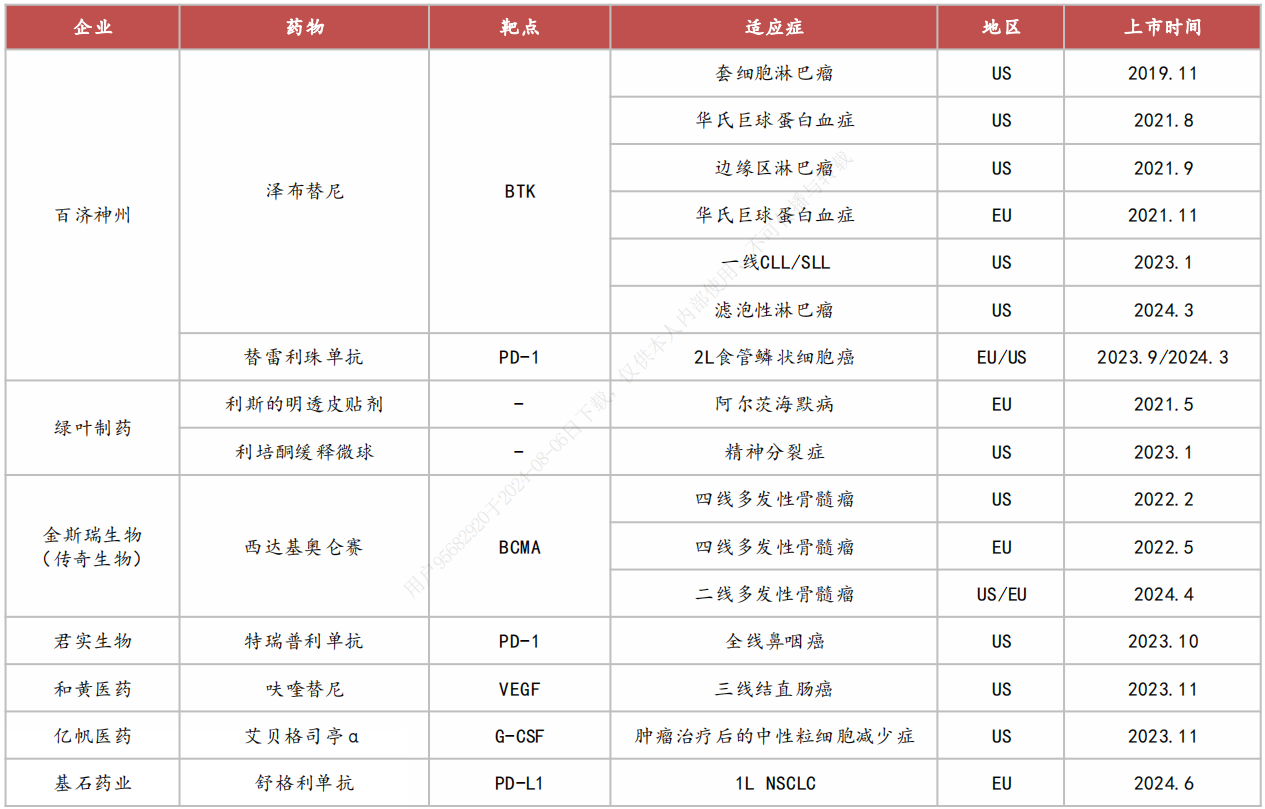
图:部分代表性的国产创新药海外上市品种
典型代表:传奇生物
2022年2月底,传奇生物旗下的西达基奥仑赛通过美国FDA批准上市,成为首款获得美国FDA批准的中国CAR-T细胞疗法;
2022年5月26日,传奇生物宣布欧盟委员会(EC)已授予西达基奥仑赛上市许可;
2024年4月22日,欧盟委员会(EC)批准西达基奥仑赛新适应症。
早在2017年12月,强生与传奇生物签订了独家全球许可和合作协议,以开发和商业化西达基奥仑赛,这笔交易首付款达3.5亿美元。在美国,两家公司的权益按50:50分成。2023年,西达基奥仑赛全球销售额达4.98亿美元。
(二)License-out
随着国内新药创制水平的不断提升,国产创新药的国际认可度稳步上升,国产创新药License-out金额持续攀升。
据不完全统计,超过30个License-out项目总交易金额超5亿美元,其中超20亿美元的项目包括科伦药业的7个ADC项目(94.7亿美元)、百利天恒的BL-B01D1(84亿美元)、康方生物AK112(50亿美元)、荣昌生物的纬迪西妥单抗(26亿)等,国产新药License-out交易金额正快速提升。
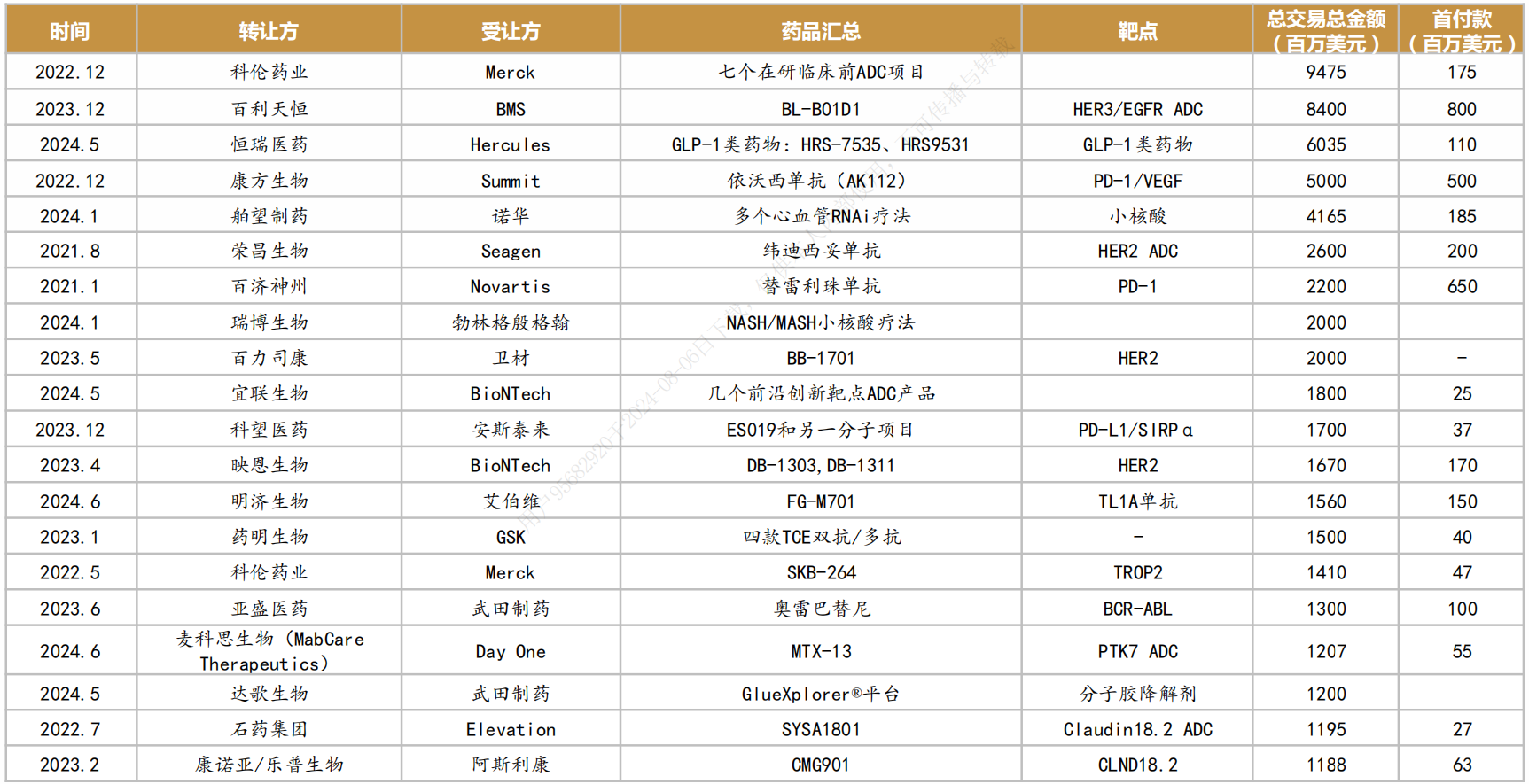
图:国产创新药License-out金额排序
随着时间的发展,传统的License-out也进化出了一些新趋势:比如国内创新药企业通过技术入股,与海外资本一起攒局,“找不到MNC(跨国药企)就先卖给中间商,没有中间商就和海外资本一起创造中间商”。尽管确实有资本运作的味道,但不能否认这种出海模式的好处,既能通过早期管线创造一定价值,又能通过股权,降低自身风险、参与公司决策,并锁定更多远期收益。
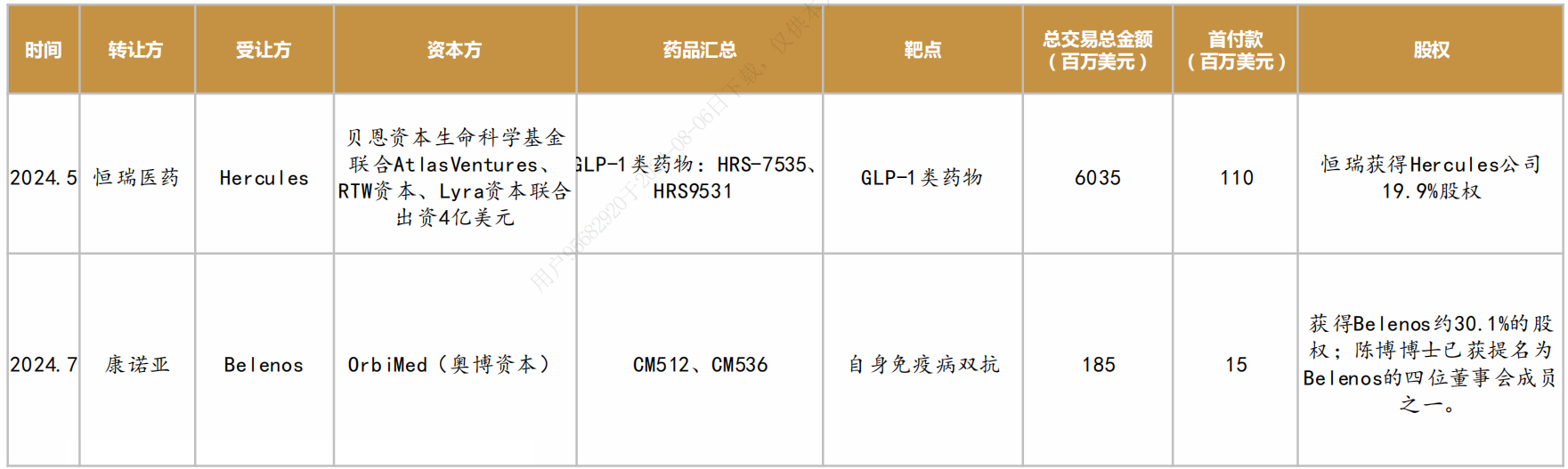
(三)FDA特殊审评通道
这里也有必要单独讲一下FDA的特殊审评通道。
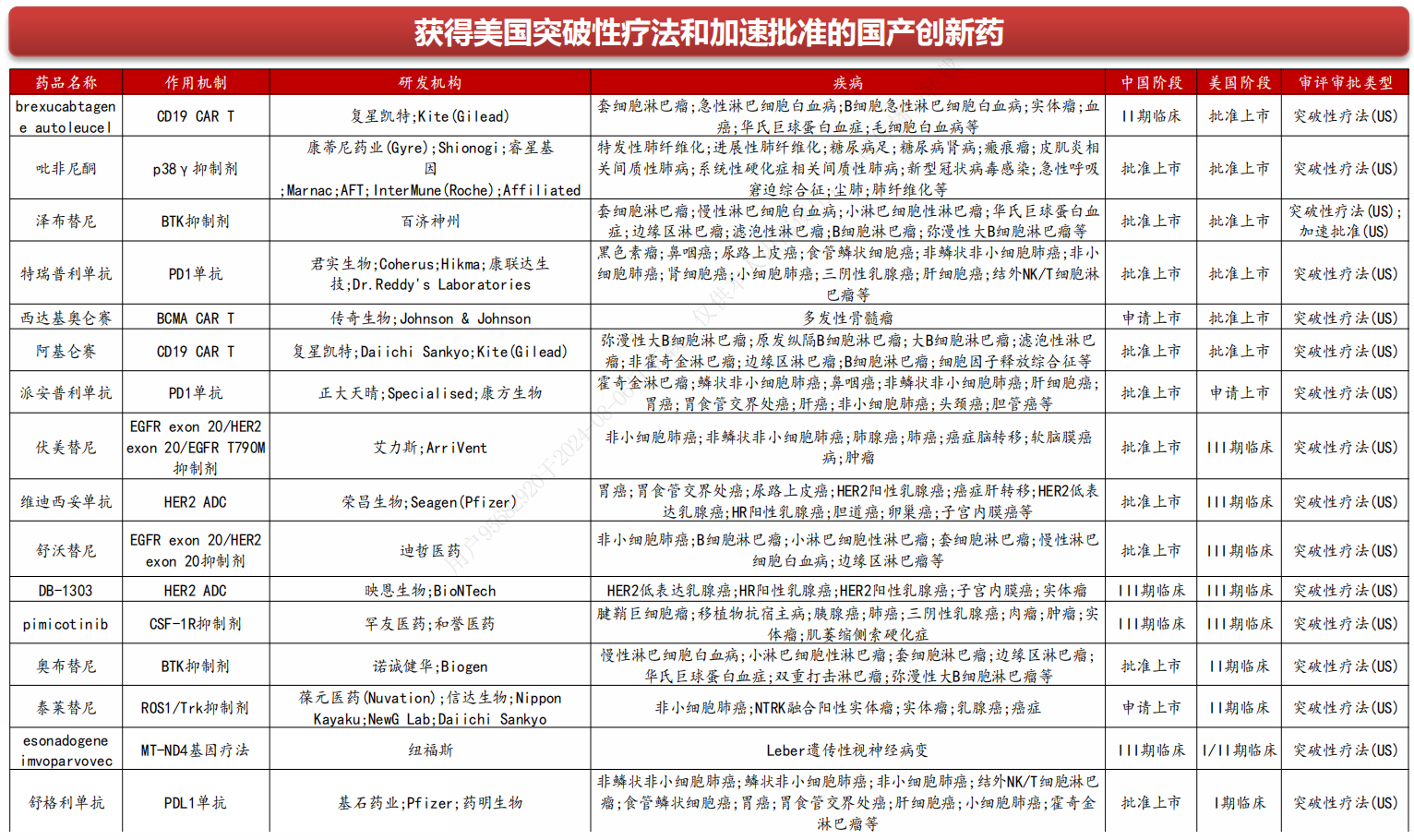
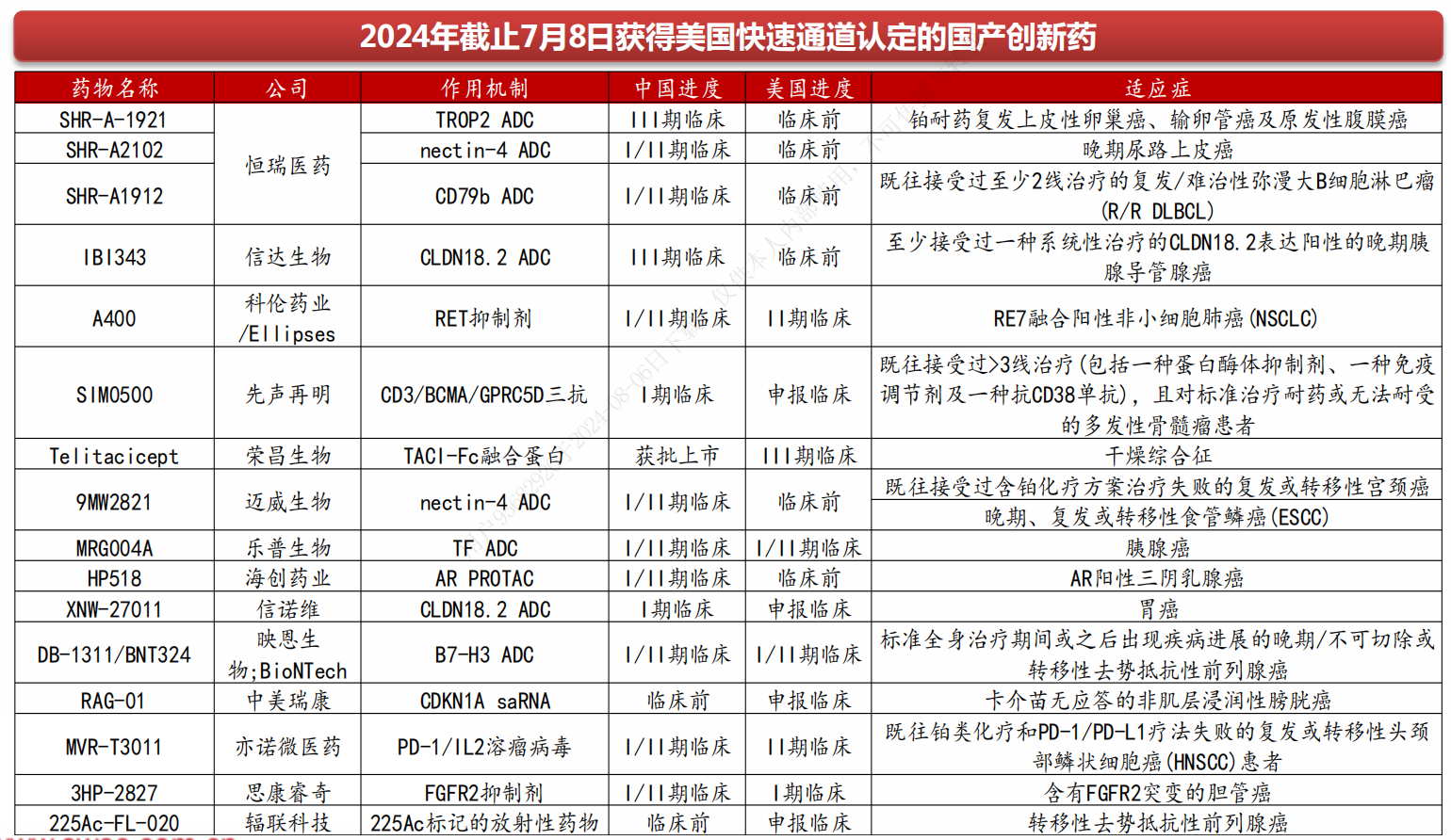
FDA为鼓励罕见病和严重疾病创新药的开发,制定了一系列的特殊审评通道,包括:孤儿药、快速通道、加速审批、优先审评和突破性疗法认定。
1)孤儿药
美国国会于1983年推出《孤儿药法案》,提出一系列推动罕见病药物研发的政策,如税收抵免(临床研发成本的25%),为孤儿药项目提供研究资助,免除上市申报注册费用(300万美元左右),上市后7年市场独占期等。此外,临床试验期间,药企还可与FDA对临床试验的设计进行紧密沟通,缩短IND(新药研究申请)和NDA(新药上市申请)的时间。
2)快速通道
美国于1988年推出快速审评通道计划,以“促进治疗严重疾病和满足未满足的医疗需求的药物的开发和加快审查”。在快速通道流程下,制药公司在I期试验后与FDA讨论II期试验设计,如果成功,II期试验(而非III期)将足以证明药物的安全性和有效性。根据该计划,FDA可以持续审查临床试验产生的证据(滚动审查)。
3)加速审批
衡量药物对患者生存的影响可能需要较长的试验时间和随访数据(特别是对于5年和10年生存率较高的癌症类型)。相比之下,替代终点(例如肿瘤缩小或进展)可以更快地观察到。因此,加速审批计划通过缩短临床试验持续时间并启用较少入组患者的试验设计来测量替代终点,从而加快了药物开发。有研究显示,获得加速审批的癌症药物比那些获得标准FDA批准的药物更早进入市场,平均提前了3.9年。
4)优先审评
标准NDA(产品研发相关非披露资料)的审核周期为10个月(2002年为12个月),而优先NDA的审核周期为6个月。与其他特殊指定不同,审查途径由FDA收到NDA和补充适应症材料后自行判定。
5)突破性疗法认定
2012年,美国国会引入了新的FDA审查途径:突破性疗法。该认定提供与FDA高级人员更早(I期)和更频繁的会议,以指导有效的药物开发和监管审查流程。此外,突破性疗法指定的药物可以获得快速通道计划的所有好处。
04
总结
综上所述,中国创新药产业自2015年以来,经过十年发展,已经初具成效,不管是国内市场还是海外市场,都是优秀创新药企必须要去拓展和突破的空间,尤其是在目前国内投融资低迷、医保控费压力较大的背景下,出海更是需要重点探索的方向。
目前来看,出海的思路转变已经完成,出海的路径和方式已经清晰,出海的能力也在一步步搭建和完善。我们有理由期待下一个十年,中国创新药的出海发展,大有可为。
免责声明:本报告中的信息所表述的意见并不构成对任何人的投资建议。任何情况下,本公司不承担以本报告为基础进行的任何投资活动所可能导致的风险。

